Check PCR primer specificity by BLASTing your primers
Five key points to keep in mind
A common challenge when designing primers is to avoid non-specific amplification. This is because non-specific amplification can lead to false positives in downstream analyses: if you are trying to amplify a gene of interest, but your primer also amplifies a different gene, then you could get a false positive result.
Similarly, you want to avoid even one primer binding to two locations in the genome. This is because mis-priming of one primer can lead to lower efficiency of your PCR reaction, and in extreme cases to false negatives.
And if you’re thinking about multiplexing, having primers that match multiple locations can lead to false positives and to false negatives.
Common primer design software such as Primer3 will check annealing and melting temperatures and avoid primer-dimers, but they do not check for non-specific amplification.
However, BLAST is a powerful tool used to compare sequences and search for similarities. It can be used to check the specificity of PCR primers. But there are five key points to keep in mind.
- Use shorter word-size to increase sensitivity
- Search the entire genome: Avoid filtering
- Be specific in the scope of your search
- Check BLAST hit coordinates
- Changing how BLAST tallies up scores
Let’s dive deeper into each of these points, before summarizing the BLAST parameters you should use in the command-line or in a graphical BLAST search tool like SequenceServer.
1. Use shorter word-size to increase sensitivity for primer BLAST
By default, BLAST searches with a word-size of 11 or even 28. This means that BLAST will only detect sequence similarity if there are at least 11 (or even 28) nucleotides of perfect identity. That would be inappropriate for checking primer specificity, because primers are typically 20 nucleotides long, and even partial matches can create mis-priming. So rather than using the default, specify -task blastn-short. This decreases the word-size to 7. Thus it increases BLAST’s ability to detect partial matches.
2. Search the entire genome: Avoid BLAST filtering of repetitive regions
You also want to be searching the entire genome. But because of filtering, that normally doesn’t happen. Thus, you want to switch off filtering of lower confidence or lower complexity regions. This is because part of your primer may sometimes hit a repetitive or ambiguous sequence.
To switch off filtering, specificy -dust no -soft_masking false. (Soft-masking ignores lowercase letters in the genome, and dust is a filter that removes highly repetitive sequences such as microsatellites or minisatellites).
3. Be specific by reducing the scope of the BLAST database you are searching with
BLAST is more sensitive if it is searching against the most appropriate database. This is because smaller databases lead to stronger (i.e., smaller) e-values.
So reduce the scope of your search. In most cases, this means searching against only your organism of interest, rather than a multi-genome database or something like “all of RefSeq” or “all of NCBI”). But if your DNA sample contains DNA from multiple organisms, as happens in symbiosis or in microbiomes, then you should use the search against that correct combination of relevant genomes.
4. Check coordinates of primer BLAST hits
Ideally, you get a single hit per primer. But if you get multiple, check their coordinates. For this, it can be helpful to download the table of all hits into excel. Make sure you also check hit orientation in comparison to query orientation - this is because BLAST doesn’t necessarily align from the first to the last nucleotide (i.e., the hit may focus on the middle part of the sequence that does match perfectly).
You should also check the distance between the extremities of the coordinates should be the size of the expected PCR product. And primer pairs want to be oriented correctly, and not too far apart (e.g., ideally under 1000 nucleotides)… if spanning too much of the genome, your PCR will likely fail.
For eukaryotes:
- If you’re amplifying genic DNA from a eukaryote, you obviously want to make sure to avoid exon-exon junctions! If your primer overlaps a splice junction, then it will struggle to anneal to genomic DNA.
- Unless an intron is very small, you likely want to avoid spanning multiple exons. Some introns span >100,000 nucleotides… Taq polymerase won’t be able to handle that during PCR!
- To check whether a gene or a spliceform is expressed, you may extract RNA and do reverse transcription into cDNA prior to PCR (i.e., RT-PCR or quantitative RT-PCR). In that case, rather than avoid exon-exon junctions, you likely want to design your primers so that they span exon-exon junctions. This should lead them to only amplify cDNA, but not any genomic DNA that might not have been fully removed during RNA extraction. Having multiple sets of such exon-exon-spanning primers can help you identify exon-skipping events, and which splice-form is expressed in which tissue.
5. Changing how BLAST tallies up scores
For primer search, some people also modify the way BLAST calculates scores… because a mismatch can severely reduce annealing. So they might add -penalty -3 -reward 1. Same thing for gaps (insertion/deletions), you might add -gapopen 5 -gapextend 2.
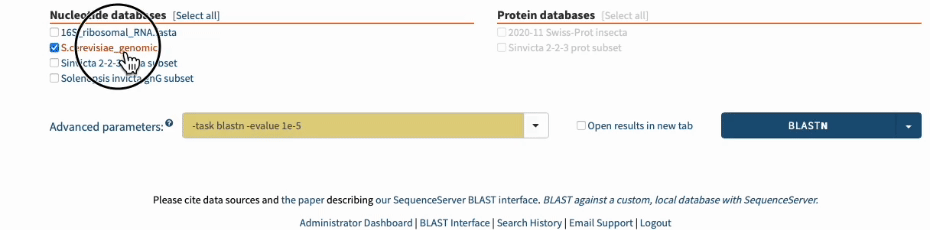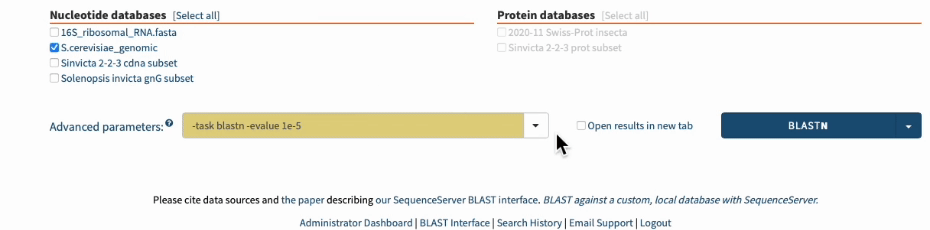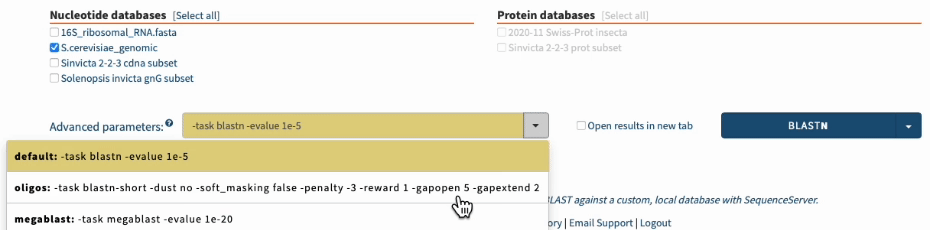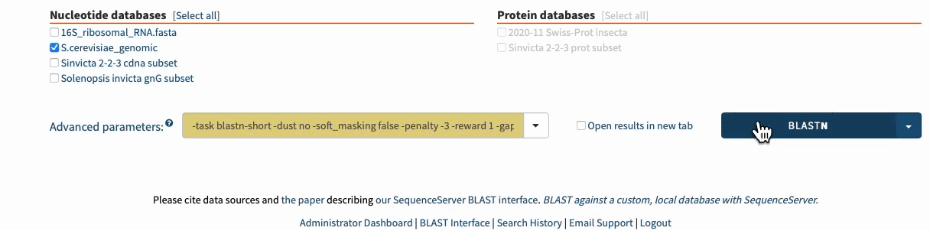
Overall, use the following options for BLASTing primer sequences:
To pull everything together, use the following BLASTN options for checking PCR primers:
-task blastn-short -dust no -soft_masking false -penalty -3 -reward 1 -gapopen 5 -gapextend 2
In the command line, you’d add those arguments to the end of the BLASTN command.
In SequenceServer, you can just select those options from the “Advanced Parameters” drop-down menu.
Going one step further by BLASTing both primers together
Above, we discussed the BLAST parameters that work great for short oligonucleotide sequences like primers (but also Illumina adapters or crispr-Cas9 guide gRNAs).
Some off-target hybridization of your primers to your genome may occur. But that really will only be problematic if both primers hybridize to the same off-target sequence. What are the odds? Well if you’re looking at recent gene duplicates, it is actually quite likely!
So you can go one step further by BLASTing both primers together. For that: concatenate the two primers together, separated by a few “NNN” nucleotides, then hit BLAST. You should get an overview such as the following:
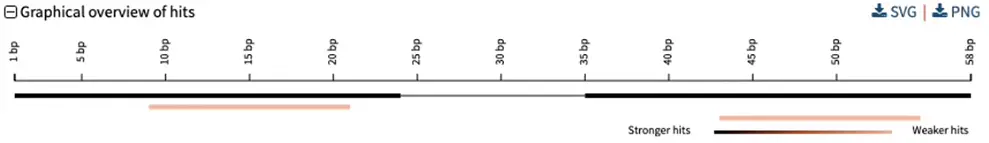
Here, a single genomic segment (the two black rectangles connected by a thin line) aligns to the contactenated primer. Each primer also has a weaker alignment to a different segment (in pink). Scrolling further down allows us to see exactly where and how the primers align to the genome:

As well as weaker secondary alignemnts (only one shown):

SequenceServer Cloud makes it easy to perform sequence search results and to interpret them. For this, it leverages cloud computing, publication-ready graphics that facilitate interpretation, and a powerful graphical interface for configuring BLAST databases.
![]()
Stay up to date
To receive the latest news from our team, enter your email: