BLAST servers for custom and standard data
Accelerating research with intuitive BLAST servers since 2011:
Join >20,000 users and experience the power of SequenceServer
Researchers and staff scientists in hundreds of companies and labs use SequenceServer to BLAST their nucleotide and amino-acid sequences. We've been cited 250 times. Applications range from vaccine development to cancer research, molecular diagnostics, synthetic biology, metagenomics, barcoding, evolutionary genomics and tracking illegal ivory seizures.
SequenceServer is the best way to BLAST custom genomes assemblies, transcriptomes and proteomes. It works great for standard databases (hg38, SwissProt, UniRef, RefSeq...) without queues or limitations. And it's wonderful for sharing and annotating data and results with your team, your students, or your community.
Organizations worldwide use SequenceServer
Benefits of using SequenceServer Cloud
Just do the research
Focus on the science, not UNIX, servers, updates or waiting for NCBI.
Simply upload FASTA files or select standard sequence databases.
Begin BLASTing within seconds.
250 citations can't be wrong.
Focus on the biology.
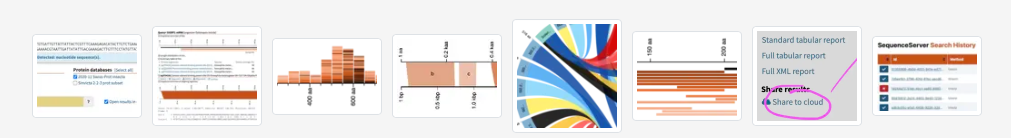
Powerful visualizations
Gain deep insight through diverse visualization approaches.
All BLAST results include pairwise alignment graphs, Circos-style overview plots, and length distribution histograms. These plots clarify and help to interpret analysis results. Publication-ready export in .PNG or .SVG formats. You work faster and more accurately.
Intelligent user interface
Faster & more accurate.
Our user interface elegantly aims to make you happy. "AI" detection of nucleotides vs amino-acids. Automagic FASTQ to FASTA conversion. Automagic selection of the best BLAST method.
Such features accelerate your workflow and reduce your chances of making errors.
Share and collaborate
Have a central SequenceServer database for your team.
Share databases and BLAST analysis results with colleagues. Foster collaboration and knowledge-sharing within your team. Annotate and comment on BLAST analyses (like in Acrobat). Share result with peers to leverage their expertise.
Export data in various formats
Streamline your R&D by integrating with other tools.
Download BLAST reports as HTML, XML or tab-delimited text. Programmatically submit and retrieve remote BLAST searches using python or command-line API. Import results into R or python. Export FASTA hit sequences or pairwise alignments. Link out to NCBI, Uniprot, jBrowse, IGV, or custom databases.
Custom links and design
Customize your SequenceServer and integrate it with other resources.
Add links from search hits to custom genome browsers or domain-specific databases. Integrate SequenceServer with other parts of your lab or community website. Change fonts, colors, and logos.
Don't melt your laptop!
SequenceServer Cloud supports intense and long-running queries.
SequenceServer Cloud is high-performance, high-availability, and scalable
BLAST platform. It won't overheat (unlike your laptop!).
Your BLAST search starts instantly. We intelligently leverage cloud computing to support large queries.
Keep your laptop for lap-top-work.
Support your students
Intuitive BLAST analysis for large student cohorts.
No more waiting at the NCBI website. We leverage cloud computing to instantly start your BLAST search. SequenceServer intelligently scales to access (almost) infinite cloud resources. Hundreds of students can run BLAST seraches simultaneously. Perfect for teaching labs with large class sizes. Our intuitive user interface and visualizations flatten learning curves. Monitor student engagement with detailed logging.
Robust, Secure and private
Secured to the higest standards.
We prioritize your data's security and privacy. With advanced SSL/TLS encryption, proactive firewalls, intrusion detection, and exclusive Virtual Private Clouds (VPCs), we safeguard your data to the highest industry standards. Control access with popular Single-Sign-On (SSO) providers and track interactions with detailed audit trail logs. Trusted by academics, biotech, pharma, and agro-industry users worldwide, we're committed to protecting your data as if it were our own.
Use Cases
Comparative Genomics
Compare genomes across multiple species, identify conserved regions, protein domains, homologous genes, copy number differences, sequence differences, species, and evolutionary relationships. Deepen our understanding of the genetic basis of various biological traits and functions, and evolutionary histories.
Gene Curation and Annotation
Validate and refine gene predictions using SequenceServer BLAST searches against known gene sequences and protein domains. Identify potential inaccuracies in gene annotations and improve qualities of genesets and genome assemblies. Graphs for visualizing pairwise alignments and hit length distributions are particularly helpful.
Design of Diagnostic Markers
SequenceServer helps identify unique sequence regions in viruses, bacteria or other target organisms that can serve as diagnostic markers for disease detection. Design or PCR or qPCR primers and probes, or specific guide RNA oligos (gRNA) for Crispr/Cas9 assays. Improving specificity and sensitivity of molecular-genomic approaches supports disease surveillance and management efforts.
Functional Characterization
Identify and analyze novel genes of interest in non-model organisms, newly sequenced or understudied genomes. By comparing these sequences to known genes in other organisms, researchers can gain insights into the potential functions and biological roles of the newly discovered genes, guiding further experimental validation and functional studies.
Peer-reviewed citations to our publication
The 2019 MBE paper in which we published SequenceServer is cited almost weekly, with more than 200 citations.
Check Google Scholar for the latest, or a slightly dated list here.
- Tinoco (2021) Ancient role of sulfakinin/cholecystokinin-type signalling in inhibitory regulation of feeding processes revealed in an echinoderm. eLife.
- Acha (2021) A Traceable DNA-Replicon Derived Vector to Speed Up Gene Editing in Potato: Interrupting Genes Related to Undesirable Postharvest Tuber Traits as an Example. Plants.
- Sakuraoka (2021) Comparative Genome Analysis Reveals Differences in Biocontrol Efficacy According to Each Individual Isolate Belonging to Rhizospheric Fluorescent Pseudomonads. Microbes and Environments.
- Guérin (2021) Transcriptome architecture and regulation at environmental transitions in flavobacteria: the case of an important fish pathogen. Nature.
- Lei (2021) TeaPGDB: Tea Plant Genome Database. Beverage Plant Research.
- Morohoshi (2021) Comparative genome analysis reveals the presence of multiple quorum-sensing systems in plant pathogenic bacterium, Erwinia rhapontici. Bioscience, Biotechnology, and Biochemistry.
- Barreira (2021) AniProtDB: A Collection of Consistently Generated Metazoan Proteomes for Comparative Genomics Studies. Molecular Biology and Evolution.
- Talamantes-Becerra (2021) omicR: A tool to facilitate BLASTn alignments for sequence data. SoftwareX.
- Wang (2021) PSDX: A comprehensive multi-omics association database of Populus trichocarpa with a focus on the secondary growth in response to stresses. Frontiers in Plant Science.
- Hu (2021) Database Resources for Functional Circular RNAs. RNA Bioinformatics.
- Zhu (2021) High‐density Linkage Map Construction and QTL Analysis of Fiber Quality and Lint Percentage in Tetraploid Cotton (Gossypium hirsutum L.) Crop Science.
- Caurcel (2021) MolluscDB: a genome and transcriptome database for molluscs. Royal Society Publishing.
- Terraneo (2021) Phylogenomics of Porites from the Arabian Peninsula. Molecular Phylogenetics and Evolution.
- Adams (2021) Rust expression browser: an open source database for simultaneous analysis of host and pathogen gene expression profiles with expVIP. BMC Genomics.
- Darragh (2021) A novel terpene synthase controls differences in anti-aphrodisiac pheromone production between closely related Heliconius butterflies. PLoS Biology.
- Miyazaki (2021) Evolutionary transition of doublesex regulation in termites and cockroaches: from sex-specific splicing to male-specific transcription. bioRxiv.
- Schön (2021) Picozoa are archaeplastids without plastid. bioRxiv.
- Kim (2021) The Lithuanian reference genome LT1-a human de novo genome assembly with short and long read sequence and Hi-C data. bioRxiv.
- Zhang (2021) NBIGV-DB: a dedicated database of non-B cell derived immunoglobulin variable region. Gene.
- Cosi (2021) StarBLAST: a scalable BLAST+ solution for the classroom. The Journal of Open Source Education.
- Garczarek (2021) Cyanorak v2. 1: a scalable information system dedicated to the visualization and expert curation of marine and brackish picocyanobacteria genomes. Nucleic Acids Research.
- Braymer (2020) Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research.
- Fu (2020) A gene prioritization method based on a swine multi-omics knowledgebase and a deep learning model. Nature.
- Zhang (2020) Molecular and functional characterization of somatostatin-type signalling in a deuterostome invertebrate. Open Biology.
- Simon (2020) Genomic, metabolic and phenotypic variability shapes ecological differentiation and intraspecies interactions of Alteromonas macleodii. usc.edu.au
- Billard (2020) A natural mutational event uncovers a life history trade-off via hormonal pleiotropy. Current Biology.
- Bock (2020) Mating Type Idiomorphs, Heterothallism, and High Genetic Diversity in Venturia carpophila, Cause of Peach Scab. Phytopathology.
- Swann (2020) The immunogenetics of sexual parasitism. Science.
- Yañez-Guerra (2020) Echinoderms provide missing link in the evolution of PrRP/sNPF-type neuropeptide signalling. eLife Sciences.
- Triant (2020) Using online tools at the Bovine Genome Database to manually annotate genes in the new reference genome. Animal Genetics.
- Li (2020) Dynamic transcriptional and chromatin accessibility landscape of medaka embryogenesis. Genome Research.
- Reichler (2020) Interventions designed to control postpasteurization contamination in high-temperature, short-time-pasteurized fluid milk processing facilities: A case study on the effect of employee training, clean-in-place chemical modification, and preventive maintenance programs. Journal of Dairy Science.
- Kim (2020) The chloroalkaloid (−)-acutumine is biosynthesized via a Fe(II)- and 2-oxoglutarate-dependent halogenase in Menispermaceae plants. Nature Communications.
- Wang (2020) Coexpression Analysis Reveals Dynamic Modules Regulating the Growth and Development of Cirri in the Rattans (Calamus simplicifolius and Daemonorops jenkinsiana). Frontiers in Genetics.
- Dallinger (2020) Metallomics reveals a persisting impact of cadmium on the evolution of metal-selective snail metallothioneins. Oxford Academic.
- Gauthier (2020) Contrasting genomic and phenotypic outcomes of hybridization between pairs of mimetic butterfly taxa across a suture zone. Molecular Ecology.
- Ainouche (2020) The repetitive content in lupin genomes. The Lupin Genome.
- Singh (2020) Overview of Genomic Resources Available for Lupins with a Focus on Narrow-Leafed Lupin (Lupinus angustifolius). The Lupin Genome.
- Salmon (2020) Abdelkader Aïnouche, Aurore Paris, Delphine Giraud, Jean Keller, Pauline Raimondeau, Frédéric Mahé, Pavel Neuman, Petr Novak, Jiri Macas, Malika Aïnouche. The Lupin Genome.
- Singh (2020) Overview of Genomic Resources Available for Lupins with a Focus on Narrow-Leafed Lupin (Lupinus angustifolius). The Lupin Genome.
- Waterhouse (2020) Characterization of Insect Immune Systems from Genomic Data. Immunity in Insects. Springer Protocols Handbooks.
- Koch (2020) Genomic, metabolic and phenotypic variability shapes ecological differentiation and intraspecies interactions of Alteromonas macleodii. Scientific Reports.
- Gui (2020) ZEAMAP, a Comprehensive Database Adapted to the Maize Multi-Omics Era. iScience.
- Moreland (2020) The Mnemiopsis Genome Project Portal: integrating new gene expression resources and improving data visualization. Database.
- Yu (2020) SAGER: a database of Symbiodiniaceae and Algal Genomic Resource. Database.
- Wu (2020) PncStress: a manually curated database of experimentally validated stress-responsive non-coding RNAs in plants. Database.
- Barreira (2020) AniProtDB: A Collection of Uniformly Generated Metazoan Proteomes for Comparative Genomics Studies bioRxiv.
- Zhang (2020) NBIGV-DB: a dedicated database of non-B cell derived immunoglobulin variable region. Gene.
- Swann (2020) The immunogenetics of sexual parasitism. Science.
- Colin (2020) The chloroalkaloid (−)-acutumine is biosynthesized via a Fe(II)- and 2-oxoglutarate-dependent halogenase in Menispermaceae plants. Nature Communications.
- Li (2020) Dynamic transcriptional and chromatin accessibility landscape of medaka embryogenesis. Genome Research.
- Yañez-Guerra (2020) Echinoderms provide missing link in the evolution of PrRP/sNPF-type neuropeptide signalling. eLife.
- Hart (2020) Visual Opsin Diversity in Sharks and Rays. Molecular Biology and Evolution.
- Gauthier (2020) Contrasting genomic and phenotypic outcomes of hybridization between pairs of mimetic butterfly taxa across a suture zone. Molecular Ecology.
- Dallinger (2020) Metallomics reveals a persisting impact of cadmium on the evolution of metal-selective snail metallothioneins. Metallomics.
- Koch (2020) Genomic, metabolic and phenotypic variability shapes ecological differentiation and intraspecies interactions of Alteromonas macleodii. Scientific Reports.
- Gui (2020) ZEAMAP, a Comprehensive Database Adapted to the Maize Multi-Omics Era. iScience.
- Wu (2020) PncStress: a manually curated database of experimentally validated stress-responsive non-coding RNAs in plants. Database.
- Moreland (2020) The Mnemiopsis Genome Project Portal: integrating new gene expression resources and improving data visualization. Database.
- Yu (2020) SAGER: a database of Symbiodiniaceae and Algal Genomic Resource. Database.
- Reichler (2020) Interventions designed to control postpasteurization contamination in high-temperature, short-time-pasteurized fluid milk processing facilities: A case study on the effect of employee training, clean-in-place chemical modification, and preventive maintenance programs. Journal of Dairy Science.
- Wang (2020) Coexpression Analysis Reveals Dynamic Modules Regulating the Growth and Development of Cirri in the Rattans (Calamus simplicifolius and Daemonorops jenkinsiana). Frontiers in Genetics.
- Triant (2020) Using online tools at the Bovine Genome Database to manually annotate genes in the new reference genome. Animal Genetics.
- Goodheart (2020) Laboratory culture of the California Sea Firefly Vargula tsujii (Ostracoda: Cypridinidae): Developing a model system for the evolution of marine bioluminescence. Scientific Reports.
- Waterhouse (2020) Characterization of Insect Immune Systems from Genomic Data. Immunity in Insects. Springer Protocols Handbooks.
- Singh (2020) Overview of Genomic Resources Available for Lupins with a Focus on Narrow-Leafed Lupin (Lupinus angustifolius). The Lupin Genome. Compendium of Plant Genomes. Springer, Cham.
- Alves (2020) Incorporating personality in user interface design: A review. Personality and Individual Differences.
- Wang et al. (2020) MaGenDB: a functional genomics hub for Malvaceae plants. Nucleic Acids Research.
- Shamimuzzaman et al. (2020) Bovine Genome Database: new annotation tools for a new reference genome. Nucleic Acids Research.
- Goodheart (2020) Laboratory culture of the California Sea Firefly Vargula tsujii (Ostracoda: Cypridinidae): Developing a model system for the evolution of marine bioluminescence. Scientific Reports.
- Thole (2019) RNA-seq, de novo transcriptome assembly and flavonoid gene analysis in 13 wild and cultivated berry fruit species with high content of phenolics. BMC Genomics.
- Prieto et al. (2019) Grape Biotechnology: Past, Present, and Future. SpringerLink.
- Sánchez (2019) Humberto Prieto, María Miccono, Carlos Aguirre. The Grape Genome.
- Elsy (2019) Xenopus laevis FGF16 activates the expression of genes coding for the transcription factors Sp5 and Sp5l. International Journal of Developmental Biology.
- Li et al. (2019) Population Genomic Signatures of Genetic Structure and Environmental Selection in the Catadromous Roughskin Sculpin Trachidermus fasciatus. Genome Biology and Evolution.
- Bublitz et al. (2019) Peptidoglycan Production by an Insect-Bacterial Mosaic. Cell.
- Lismont (2019) Using BRET-based G protein biosensors to unravel the G protein mediated pathways in insects. KU Leuven.
- Liou (2019) Enzyme structure, function, and evolution in flavonoid biosynthesis. Massachusetts Institute of Technology, Department of Biology.
- Botwright (2019) Greenlip Abalone (Haliotis laevigata) Genome and Protein Analysis Provides Insights into Maturation and Spawning. G3 Genes | Genomes | Genetics.
- Almansoura and Alhagrib (2019) MMRdb: Measles, mumps, and rubella viruses database and analysis resource. Infection, Genetics and Evolution.
- Barbosa et al. (2019) ParaDB: A manually curated database containing genomic annotation for the human pathogenic fungi Paracoccidioides spp. PLoS Neglected Tropical Disease.
- Levsh (2019) Independent evolution of rosmarinic acid biosynthesis in two sister families under the Lamiids clade of flowering plants. Journal of Biological Chemistry.
- Ravindran et al. (2019) Daphnia stressor database: Taking advantage of a decade of Daphnia ‘-omics’ data for gene annotation . Scientific Reports.
- Mirdita et al. (2019) MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics.
- Li et al. (2019) First mitochondrial genome of a periwinkle from the genus Littoraria: Littoraria sinensis. Mitochondrial DNA Part B
- Chen et al. (2019) Neuropeptide precursors and neuropeptides in the sea cucumber Apostichopus japonicus: a genomic, transcriptomic and proteomic analysis. Scientific Reports.
- Wagner et al. (2019) The ectomycorrhizospheric habitat of Norway spruce and Tricholoma vaccinum: Promotion of plant growth and fitness by a rich microorganismic community. Frontiers in Microbiology.
- Rungrat et al. (2019) A genome-wide association study of non-photochemical quenching in response to local seasonal climates in Arabidopsis thaliana. Plant Direct.
- Bizzarri et al. (2019) Interplay of chimeric mating-type loci impairs fertility rescue and accounts for intra-strain variability in Zygosaccharomyces rouxii interspecies hybrid ATCC42981. Frontiers in Genetics.
- Nowicki et al. (2019) Taraxacum kok-saghyz (rubber dandelion) genomic microsatellite loci reveal modest genetic diversity and cross-amplify broadly to related species. Scientific Reports.
- Liang et al. (2019) Developmental expression and evolution of hexamerin and haemocyanin from Folsomia candida (Collembola). Insect Molecular Biology.
- Meng et al. (2019) CircFunBase: a database for functional circular RNAs. Database.
- Moghul et al. (2019) Choosing the best gene predictions with GeneValidator. Kollmar M. (eds) Gene Prediction. Methods in Molecular Biology, vol 1962.
- Brandies et al. (2018) Disentangling the mechanisms of mate choice in a captive koala population. PeerJ.
- Young et al. (2018) Evidence for sexual reproduction: Identification, frequency, and spatial distribution of Venturia effusa (pecan scab) mating type idiomorphs Ecology and Epidemiology.
- Reichler et al. (2018) Pseudomonas fluorescens group bacterial strains are responsible for repeat and sporadic postpasteurization contamination and reduced fluid milk shelf life. Journal of Dairy Science.
- Torrens-Spence et al. (2018) Complete pathway elucidation and heterologous reconstitution of Rhodiola salidroside biosynthesis. Molecular Plant.
- Lamb et al. (2018) Evolution of the shut-off steps of vertebrate phototransduction. Open Biology.
- Elsik et al. (2018) Hymenoptera Genome Database: Using HymenopteraMine to enhance genomic studies of hymenopteran insects. Eukaryotic Genomic Databases, Methods in Molecular Biology.
- Hagen et al. (2018) Bovine Genome Database: Tools for mining the Bos taurus genome. Eukaryotic Genomic Databases, Methods in Molecular Biology.
- Blanchoud et al. (2018) De novo draft assembly of the Botrylloides leachii genome provides further insight into tunicate evolution . Scientific Reports.
- Fallon et al. (2018) Firefly genomes illuminate parallel origins of bioluminescence in beetles. eLife.
- Sanders et al. (2018) FusoPortal: An interactive repository of hybrid MinION-sequenced Fusobacterium genomes improves gene identification and characterization. mSphere.
- Dvorak et al. (2018) Metal binding functions of metallothioneins in the slug Arion vulgaris differ from metal-specific isoforms of terrestrial snails. Metallomics.
- Chen et al. (2018) MGH: a genome hub for the medicinal plant maca (Lepidium meyenii). Database.
- Borrell et al. (2018) Genetic diversity maintained among fragmented populations of a tree undergoing range contraction. Heredity.
- Torrens-Spence et al. (2018) Monoamine biosynthesis via a noncanonical calcium-activatable aromatic amino acid decarboxylase in psilocybin mushroom. ACS Chemical Biology.
- Tangherlini et al. (2018) GLOSSary: The global ocean 16S subunit web accessible resource BMC Bioinformatics.
- Challis et al. (2017) GenomeHubs: Simple containerized setup of a custom Ensembl database and web server for any species. Database.
- Kim et al. (2017) The genome of the freshwater monogonont rotifer Brachionus calyciflorus. Molecular Ecology Resources
- Naas et al. (2017) Beta-lactamase database (BLDB) – structure and function Journal of Enzyme Inhibition and Medicinal Chemistry.
- Seim et al. (2017) Whole-Genome Sequence of the Metastatic PC3 and LNCaP Human Prostate Cancer Cell Lines. G3: Genes, Genomes, Genetics.
- Pracana et al. (2017) Fire ant social chromosomes: Differences in number, sequence and expression of odorant binding proteins. Evolution Letters.
- Suwansa-ard et al. (2017) Transcriptomic discovery and comparative analysis of neuropeptide precursors in sea cucumbers (Holothuroidea). Peptides.
- Carson et al. (2017) Bacteriocins of non-aureus staphylococci isolated from bovine milk. Applied and Environmental Microbiology.
- Shockey et al. (2017) Naturally occurring high oleic acid cottonseed oil: identification and functional analysis of a mutant allele of Gossypium barbadense fatty acid desaturase-2. Planta.
- Hill et al. (2017) HopBase: a unified resource for Humulus genomics. Database.
- Torres et al. (2017) LeishDB: a database of coding gene annotation and non-coding RNAs in Leishmania braziliensis. Database.
- Zhu et al. (2017) CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biology.
- Wang et al. (2017) QTL mapping of selenium content using a RIL population in wheat BMC Plant Biology.
- Urban et al. (2016) PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database Scientific Reports.
- Mun et al. (2016) Lotus Base: An integrated information portal for the model legume Lotus japonicus. Scientific Reports.
- Shen et al. (2016) Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3: Genes, Genomes, Genetics
- McCormick et al. (2016) 3D sorghum reconstructions from depth images enable identification of quantitative trait loci regulating shoot architecture. bioRxiv
- Liew et al. (2016) Reefgenomics.Org - a repository for marine genomics data. Database.
- Louro et al. (2016) Having a BLAST: Searchable transcriptome resources for the gilthead sea bream and the European sea bass. Marine Genomics.
- Torrens-Spence et al. (2016) A Workflow for Studying Specialized Metabolism in Nonmodel Eukaryotic Organisms. Methods in Enzymology.
- Nakagawa et al. (2016) gEVE: a genome-based endogenous viral element database provides comprehensive viral protein-coding sequences in mammalian genomes. Database.
- Challis et al. (2016) Lepbase: the Lepidopteran genome database. bioRxiv.
- Hane et al. (2016) A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: Insights into plant‐microbe interactions and legume evolution. Plant Biotechnology Journal.
- Fu et al. (2016) Dissection of early transcriptional responses to water stress in Arundo donax L. by unigene-based RNA-seq. Biotechnology for Biofuels.
- Semmens et al. (2016) Transcriptomic identification of starfish neuropeptide precursors yields new insights into neuropeptide evolution. Open Biology.
- Page et al. (2016) blastjs: a BLAST+ wrapper for Node.js. BMC Research Notes.
- Seim et al. (2016) Multi-species sequence comparison reveals conservation of ghrelin gene-derived splice variants encoding a truncated ghrelin peptide. Endocrine.
- Janies et al. (2016) EchinoDB, an application for comparative transcriptomics of deeply-sampled clades of echinoderms. BMC Bioinformatics.
- Bernonville, Foureau, Parage et al. (2015) Characterization of a second secologanin synthase isoform producing both secologanin and secoxyloganin allows enhanced de novo assembly of a Catharanthus roseus transcriptome. BMC Genomics.
- Castro et al. (2015) Identification and heterologous expression of the chaxamycin biosynthesis gene cluster from Streptomyces leeuwenhoekii. Applied and Environmental Microbiology.
- Semmens et al. (2015) Discovery of sea urchin NGFFFamide receptor unites a bilaterian neuropeptide family. Royal Society Publishing. Open Biology.
- Petrillo et al. (2015) JRC GMO-Amplicons: a collection of nucleic acid sequences related to genetically modified organisms. Database.
- Seim et al. (2015) Comparative analysis reveals loss of the appetite-regulating peptide hormone ghrelin in falcons. General and Comparative Endocrinology.
- Elsik et al. (2015) Hymenoptera Genome Database: integrating genome annotations in HymenopteraMine. Nucleic Acids Research.
- Brandl et al. (2015) PlanMine – a mineable resource of planarian biology and biodiversity. Nucleic Acids Research.
- Kirmitzoglou I (2015) LCR-eXXXplorer: a web platform to search, visualize and share data for low complexity regions in protein sequences. Bioinformatics.
- Elphick MR (2015) Reconstructing SALMFamide neuropeptide precursor evolution in the phylum Echinodermata: ophiuroid and crinoid sequence data provide new insights. Frontiers in Endocrinology.
- Gupta Y et al. (2015) De novo assembly and characterization of transcriptomes of early-stage fruit from two genotypes of Annona squamosa L. with contrast in seed number . BMC Genomics.
- Rodrigues M (2014) Molecular biology approaches in bioadhesion research. Beilstein Journal of Nanotechnology.
- Sharma P (2014) WImpiBLAST: Web Interface for mpiBLAST to Help Biologists Perform Large-Scale Annotation Using High Performance Computing. PLoS ONE.
- Mondav R et al. (2014) Discovery of a novel methanogen prevalent in thawing permafrost. Nature Communications.
- Rowe ML et al. (2014) Neuropeptides and polypeptide hormones in echinoderms: New insights from analysis of the transcriptome of the sea cucumber Apostichopus japonicus. General and Comparative Endocrinology.
- Chiara M et al. (2013) De novo assembly of the transcriptome of the non-model plant Streptocarpus rexii employing a novel heuristic to recover locus-specific transcript clusters . PLoS ONE.
- Semmens DC et al. (2013) Discovery of a novel neurophysin-associated neuropeptide that triggers cardiac stomach contraction and retraction in starfish. Journal of Experimental Biology.
- Chiu JC et al. (2013) Genome of Drosophila suzukii, the Spotted Wing Drosophila. G3.
- Shreve J et al. (2013) A genome-wide survey of small interfering RNA and microRNA pathway genes in a galling insect. Journal of Insect Physiology.
- Berlamino et al. (2013) SymGRASS: a database of sugarcane orthologous genes involved in arbuscular mycorrhiza and root nodule symbiosis. BMC Bioinformatics.
- Elphick MR et al. (2013) The evolution and diversity of SALMFamide neuropeptides. PLoS ONE.

Always Open Source
We take pride in keeping our core open source and free for all. Explore our documentation, connect with us on our community support forum, or contribute to the GitHub repository. Together, we're advancing bioinformatics one BLAST at a time.
Ready to Revolutionize Your Sequence Analysis?
Get started with SequenceServer today
Try our Cloud ServiceLatest News
We’ve launched a referral scheme! Help your colleagues and support continued improvements to your favorite BLAST interface.
Earn up to $100 per colleague or team that signs up.
2023-10
We’ve released SequenceServer 2.2.0! It’s faster. It’s more stable. It’s more flexible.
Check the details.
2023-10
SequenceServer 2.1 is released. Many new features and improvements.
Check the full SequenceServer BLAST release notes.
2023-08